RNA-Seq - introducing Galaxy
I’m actually attending a lecture with the great name RNA-Seq, dealing with next generation sequencing (NGS). I think the lecture is more or less addressed to biological scientist and people who are working with genome analyzers, but I think there is no harm in visiting this lecture and to get to know the biologists point of view.
These scientists are using different sequencing platforms. Some popular examples are Roche 454, Illumina/Solexa, ABI SOLiD, Pacific Biosciences PacBio RS, Helicos HeliScope™ Single Molecule Sequencer or Polonator, but there are much of more such platforms. If you are interested in these different techniques, you are referred to [Met09]. There is no standard, so all these machines produce output in different formats and quality. In general you’ll get a fastq file as result of sequencing. This file contains roughly more or less small reads of sequences and a quality score of each recognized nucleotide. The quality score is encoded in ASCII characters and contains four line types. Here is an example of such a file:
@SRR039651.1 HWUSI-EAS291:8:1:1:356
TTTTGGTTTTANTTTTTAATAGGTAAATNNNNNNNT
+
BCCBAABCCC=!/=BCABB%%%%%%%%%!!!!!!!%
@SRR039651.2 HWUSI-EAS291:8:1:1:410
TGGTTTGGTTGNTATTGTGATGTATTTANNNNNNNT
+
BBB?@BBB@A0!0<B?.4B?BA?%%%%%!!!!!!!%
@SRR039651.3 HWUSI-EAS291:8:1:1:1018
TTAGTAGTGTTNGTAGAATTTTATTTGTNNNNNNNT
+
BBBB;AB?B@=!,5@B=@ABBB=B%%%%!!!!!!!%As you can see, in general the file contains an identifier line, starting with @ , the recognized sequence, a comment, starting with + , followed by the quality score for each base. It’s a big problem that there is no common standard for these quality scores, they differ in domain depending on the sequencing platform. So the original Sanger format uses PHRED scores ([EG98] and [EHWG98]) in an ASCII range 33-126 ( ! - ~ ), Solexa uses Solexa scores encoded in ASCII range 59-126 ( ; - ~ ) and with Illumina 1.3+ they introdused PHRED scores in an ASCII range 64-126 ( @ - ~ ). So you sometimes won’t be able to determine which format your fastq file comes from, the Illumina scores can be observed by all of this three example. If you want to learn more about fastq files and formats you are referred to [CFGHR10].
Interested readers are free to translate the ASCII coded quality scores of my small example to numerical quality scores and post the solution to the comment!
There is a great tool established to work with these resulting fastq files (this is just a small field of application): Galaxy. It is completely open source and written in Python. Those who already worked with it told me that you can easily extend it with plug-ins. You can choose wheter to run your own copy of this tool or to use the web platform of the Penn State. There’s a very huge ensemble of tools, I just worked with a small set of it, but I like it. It seems that you are able to upload unlimited size of data and it will never get deleted!? Not bad guys! You can share your data and working history and you can create workflows to automatize some jobs. Of course I’m excited to write an en- and decoder for other data like videos or music to and from fastq - let’s see if there’s some time ;-)
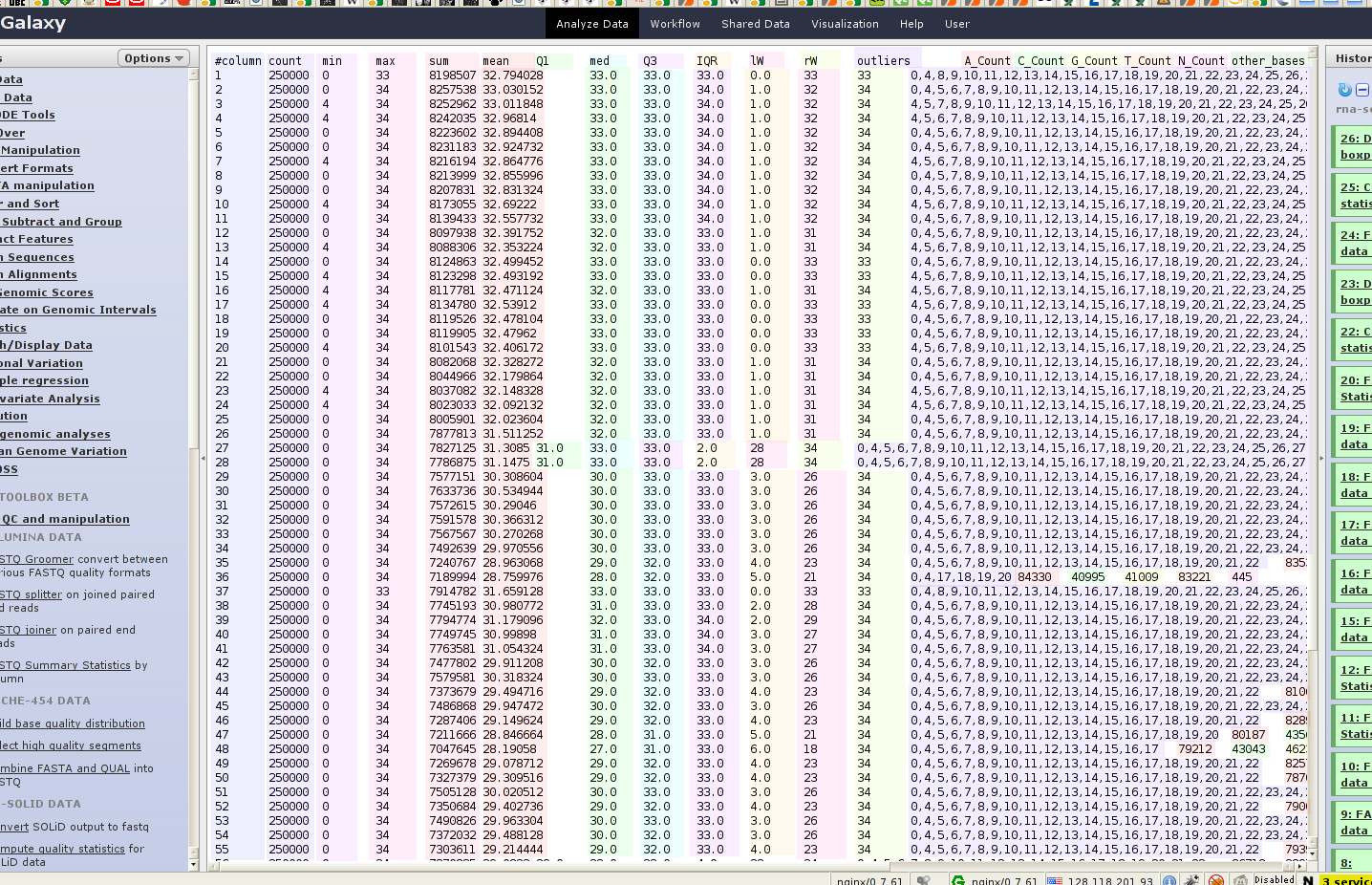
But this platform also has some inelegance’s. So there is often raw data presented in an raw format. Have a look at figure 1, you can see there is a table, columns are separated by tabs, but if one word in a column is much smaller/shorter as another one in this column this table looses the human readability! Here I’ve colorized the columns, but if the background is completely white, you have no chance to read it.
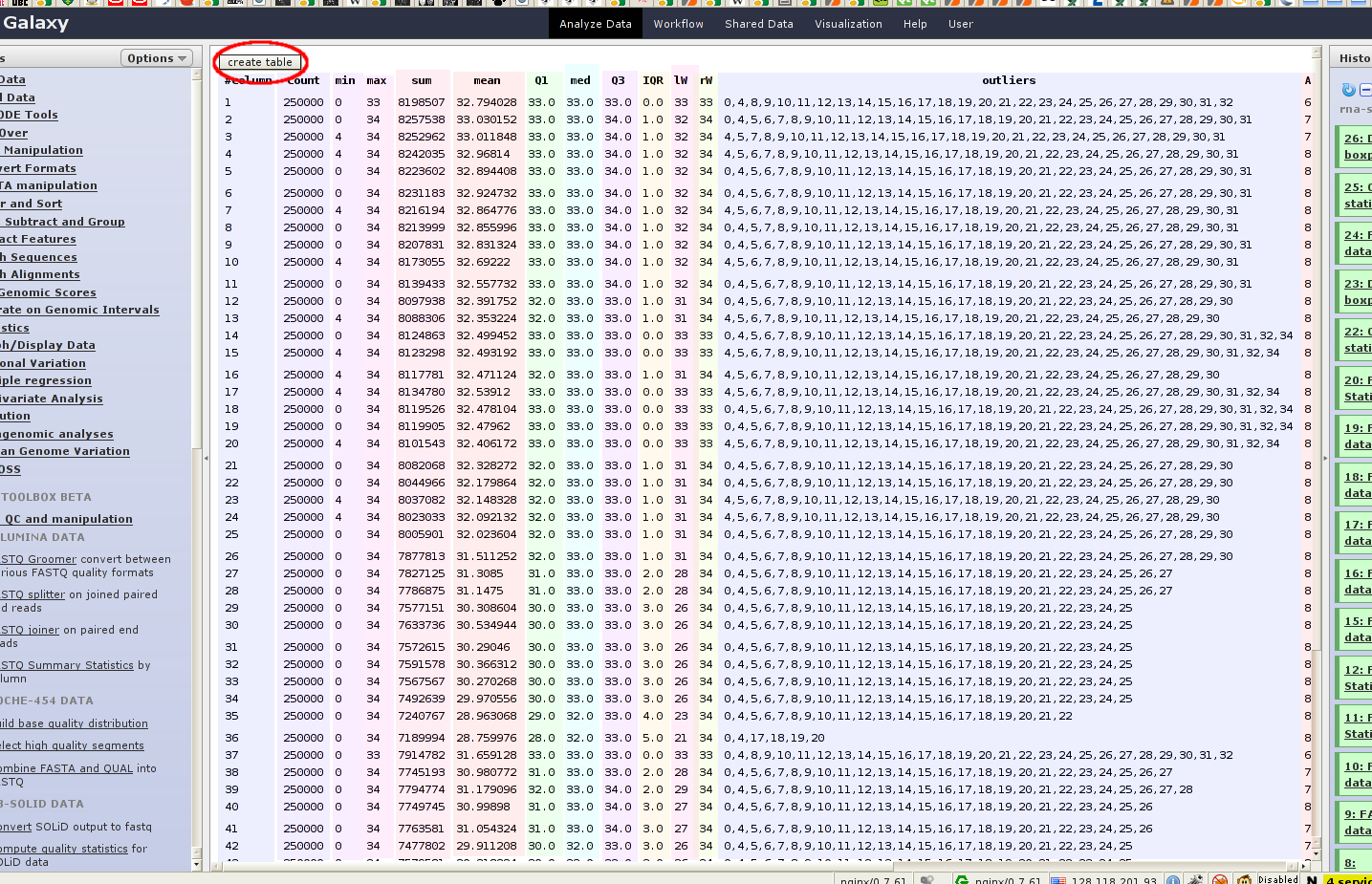
So instead of getting angry I immediately wrote a user-script. It adds a button on the top of pages with raw data and if it is clicked, it creates an HTML table of this data. You can see a resulting table in figure 2. If you think it is nice, just download it at the end of this article.
All in all I just can estimate what’s coming next!
References
- [CFGHR10]
- Peter J. A. Cock, Christopher J. Fields, Naohisa Goto, Michael L. Heuer, and Peter M. Rice. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research, 38(6):1767–1771, April 2010. http://nar.oxfordjournals.org/content/38/6/1767.abstract
- [EG98]
- Brent Ewing and Phil Green. Base-Calling of Automated Sequencer Traces Using Phred. II. Error Probabilities. Genome Research, 8(3):186–194, March 1998. http://www.ncbi.nlm.nih.gov/pubmed/9521922
- [EHWG98]
- Brent Ewing, LaDeana Hillier, Michael C. Wendl, and Phil Green. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Research, 8(3):175–185, March 1998. http://www.ncbi.nlm.nih.gov/pubmed/9521921
- [Met09]
- Michael L. Metzker. Sequencing technologies — the next generation. Nature Reviews Genetics, 11(1):31–46, December 2009. http://www.nature.com/nrg/journal/v11/n1/full/nrg2626.html

Leave a comment
There are multiple options to leave a comment: