Revived iso2l
Someone informed me about a serious bug, so I spent the last few days with rebuilding the iso2l.
This tool was a project in 2007, the beginning of my programming experiences. Not bad I think, but nowadays a pharmacist told me that there is a serious bug. It works very well on small compounds, but the result is wrong for bigger molecules.. Publishing software known to fail is not that nice and may cause serious problems, so I took some time to look into our old code…
Let me explain the problem on a little example. Let’s denote the atom Carbon \(\text{C}\) has two isotopes. The first one \(\text{C}^{12}\) with a mass of 12 and an abundance of 0.9 and another one \(\text{C}^{13}\) with a mass of 13 and an abundance of 0.1 (these numbers are only for demonstration). Let’s further assume a molecule \(\text{C}_{30}\) consisting of 30 Carbon atoms. Since we can choose from two isotopes for each position in this molecule and each position is independent from the others there are many many possibilities to create this molecule from these two isotopes, exactly \(2^{30}=1073741824\). Let’s for example say we are only using 15 isotopes of \(\text{C}^{12}\) and 15 isotopes of \(\text{C}^{13}\), there still exists \(\frac{30!}{15!\cdot 2} = 155117520\) combinations of these elements. Each combination has an abundance of \(0.9^{15} \cdot 0.1^{15} \approx 2.058911 \cdot 10^{-16}\).
Unfortunately in the first version we created a tree to calculate the isotopic distribution (here it would be a binary tree with a depth of 10). If a branch has an abundance smaller than a threshold it’s cut to decrease the number of calculations and the number of numeric instabilities. If this thresh is here \(10^{-10}\) none of these combinations would give us a peak, but if you add them all together (they all have the same mass of \(15 \cdot 12 + 15 \cdot 13 = 375\)) there would be a peak with this mass and an abundance of \(\frac{30!}{15!\cdot 2} \cdot 0.9^{15} \cdot 0.1^{15} \approx 3.193732 \cdot 10^{-8} > 10^{-10}\) above the threshold. Not only the threshold is killing peaks. This problem is only shifted if you decrease the thresh or remove it, because you’ll run in abundances that aren’t representable for your machine, especially in larger compounds (number of carbons > 100). So I had to improve this and rejected this tree-approach.
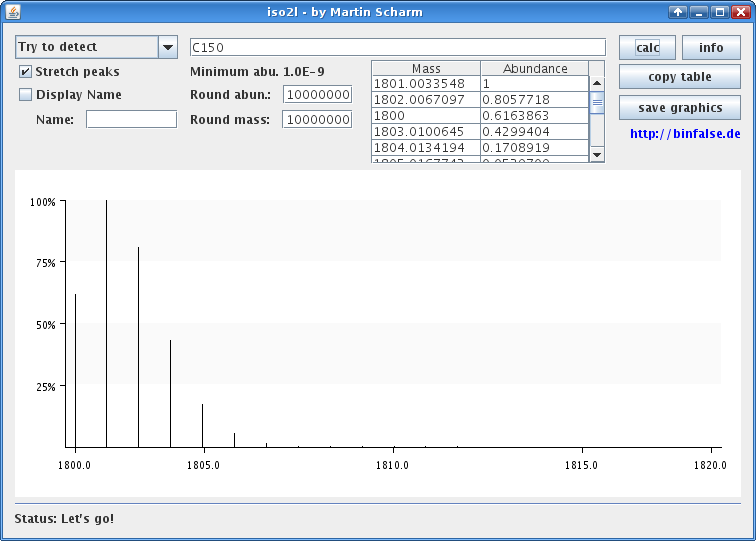
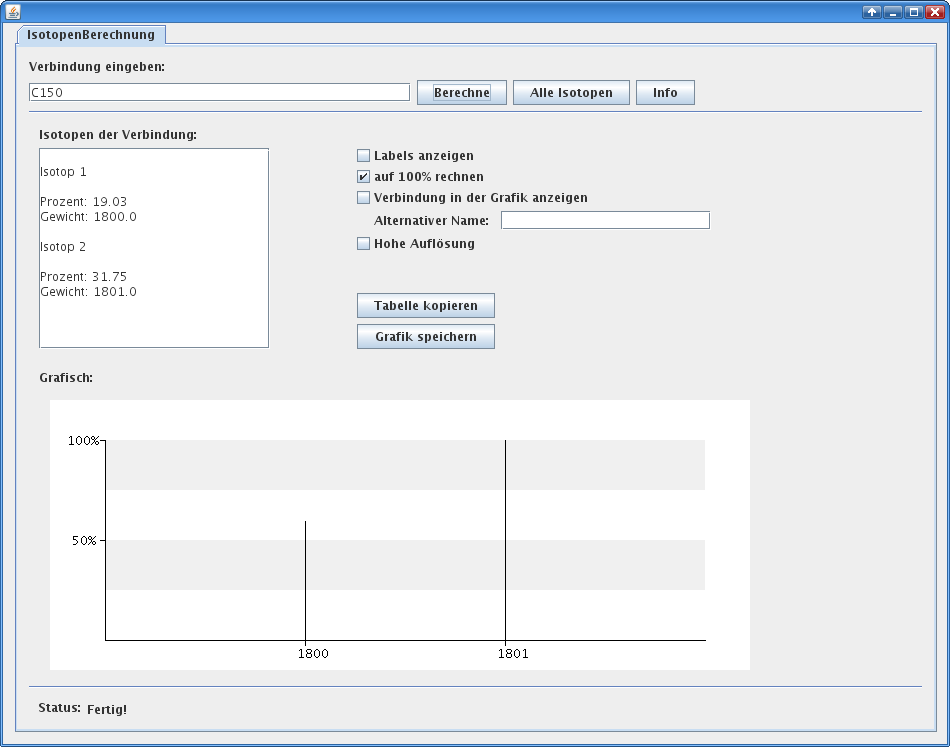
And while I’m touching the code, I translated the tool to English, increased the isotopic accuracy and added some more features. In figure 1 you can see the output of version 1 for \(\text{C}_{150}\). As I told this version loses many peaks. I contrast you can see the output of version 2 for the same compound in figure 2. Here the real isotopic cluster is visible.
Please try it out and tell me if you find further bugs or space for improvements or extensions!

Martin Scharm
Leave a comment
There are multiple options to leave a comment:
- send me an email
- submit a comment through the feedback page (anonymously via TOR)
- Fork this repo at GitHub, add your comment to the _data/comments directory and send me a pull request
- Fill the following form and Staticman will automagically create a pull request for you:
1 comment
This was a FBI Backdor(!), maybe …. . Have a deeper look at your prog-team, especially the women ;).
Have a good new year without back,trap,jump,invisible,closed,unstraight-doors.
bye