binfalse
Too much at once
December 15th, 2010Just installed a new Grml system, annoyingly from a bit too far outdated image so aptitude fails to handle everything at once…
Here is the error:
Reading package fields... 52%/usr/lib/ruby/1.8/debian/utils.rb:47:in 'pipe': Too many open files (Errno::EMFILE)
from /usr/lib/ruby/1.8/debian/utils.rb:47:in 'pipeline'
from /usr/lib/ruby/1.8/debian/utils.rb:86:in 'tar'
from /usr/lib/ruby/1.8/debian.rb:142:in 'load'
from /usr/lib/ruby/1.8/debian/utils.rb:75:in 'gunzip'
from /usr/lib/ruby/1.8/debian/utils.rb:40:in 'pipeline'
from /usr/lib/ruby/1.8/debian/utils.rb:72:in 'gunzip'
from /usr/lib/ruby/1.8/debian.rb:141:in 'load'
from /usr/lib/ruby/1.8/debian/ar.rb:150:in 'open'
from /usr/lib/ruby/1.8/debian/ar.rb:147:in 'each'
from /usr/lib/ruby/1.8/debian/ar.rb:147:in 'open'
from /usr/lib/ruby/1.8/debian.rb:140:in 'load'
from /usr/lib/ruby/1.8/debian.rb:82:in 'field'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:733:in 'field'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:751:in 'create'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:743:in 'each_index'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:743:in 'create'
from /usr/sbin/apt-listbugs:323
/usr/lib/ruby/1.8/debian.rb:198:in 'parseFields': E: required field Package not found in (Debian::FieldError)
from /usr/lib/ruby/1.8/debian.rb:196:in 'each'
from /usr/lib/ruby/1.8/debian.rb:196:in 'parseFields'
from /usr/lib/ruby/1.8/debian.rb:439:in 'initialize'
from /usr/lib/ruby/1.8/debian.rb:150:in 'new'
from /usr/lib/ruby/1.8/debian.rb:150:in 'load'
from /usr/lib/ruby/1.8/debian.rb:82:in 'field'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:733:in 'field'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:751:in 'create'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:743:in 'each_index'
from /usr/share/apt-listbugs/apt-listbugs/logic.rb:743:in 'create'
from /usr/sbin/apt-listbugs:323
E: Failed to fetch http://cdn.debian.net/debian/pool/main/k/krb5/libgssrpc4_1.8.3+dfsg-4_i386.deb: 404 Not Found
E: Sub-process /usr/sbin/apt-listbugs apt || exit 10 returned an error code (10)
E: Failure running script /usr/sbin/apt-listbugs apt || exit 10
A package failed to install. Trying to recover:
Press return to continue.Aha, too many open files.. So I had to install everything piecewise in a disturbing manner..
Btw. updating iptables 1.4.6-2 -> 1.4.10-1 before xtables-addons-common 1.23-1 -> 1.26-2 is a bad idea and fails for some reasons. So try to do it the other way round.
Crypto off
December 7th, 2010if you haven’t noticed yet: SSL is turned off…
Of course it isn’t really turned off, all content is still available through encrypted connections (all links are still working), but it’s disabled by default.
But why!?
I got a lot of mails during the last weeks, telling me that there is a problem with my SSL cert.
Yes, your browser is completely right, my cert isn’t valid because I’ve signed it by myself.. To get a trusted certificate that your browser recognizes to be valid is very expensive. For a cheap one I still have to pay about $100, that’s neither worthy nor affordable for me and my private blog. But I’m always interested in ideally offering secure mechanisms, so I tried to provide SSL.
Another reason for SSL was my auth stuff. Wordpress doesn’t provide both SSL and SSL-free access. In an installation you have to decide whether to use https://... or http://... for URL’s. So all links are either to SSL encrypted content or the next click is unencrypted. Don’t ask me why they don’t check whether SSL was turned on/off for the last query and decide afterwards on using SSL for all further links.. However, I didn’t want to authenticate myself unencrypted and so I enabled SSL by default.
To be congenial to my visitors I turned off SSL, until somebody sponsors a valid certificate. There are also many disgusting tools having problems with my website, so it might be the better way to deliver unencrypted contend. The information on my site isn’t that secret ;-)
As a consequences you aren’t able to register/login anymore. I scripted a little bit to find a secure way for authenticating myself, but you aren’t allowed to take this path :-P Nevertheless, comments are still open and doesn’t require any authentication.
If you can find any SSL zombies please inform me!
ShortCut[siblings]: tail and its derivatives
December 6th, 2010Every text-tool-user should know about tail! You can print the last few lines of a file or watch it growing. But there are three improved derivatives, just get into it.
I think there is no need for further explanation of tail itself, so lets begin with the first derivative.
colortail
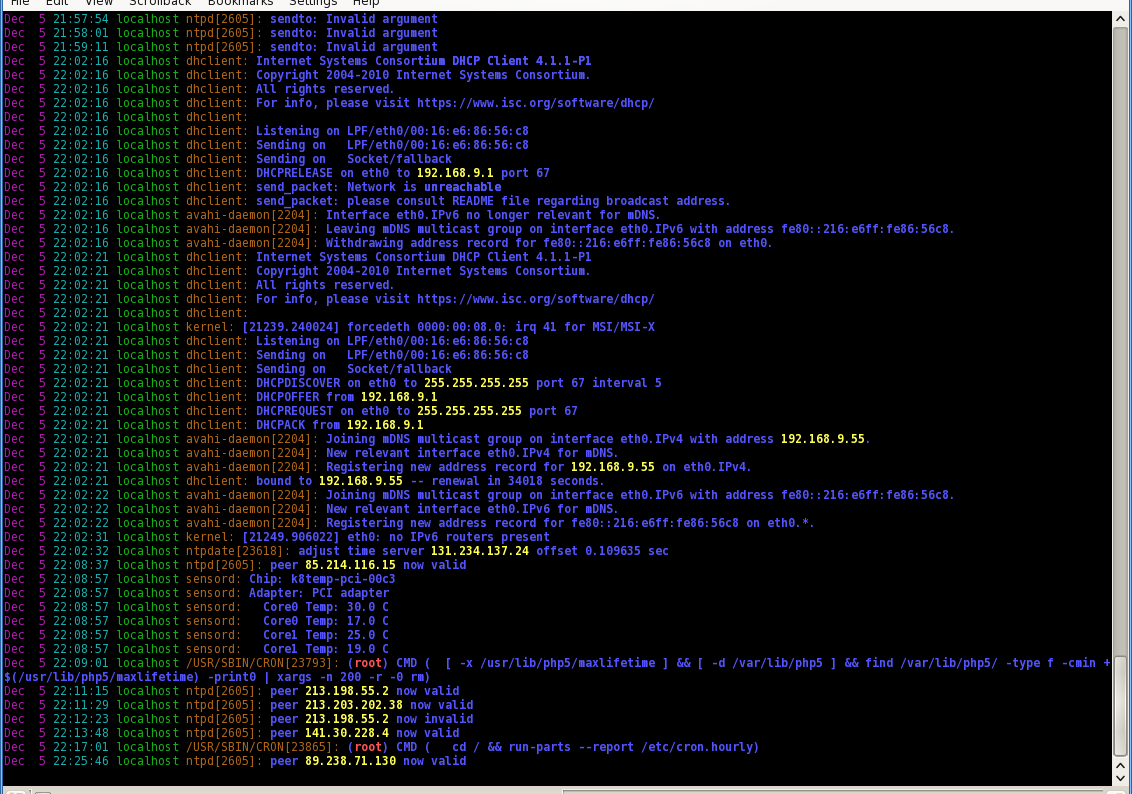
colortail is based on tail with support for colors, so it helps to keep track of important content. Common options and parameters are resembled closely to them of tail, so it won’t be a big adjustment to new circumstances for tail fans. The content that it presents is of course the same as if it comes from tail, but colorized ;)
With -k you can additional submit a configuration file that defines some regular expressions and its colors. On a Debian some examples can be found in /usr/share/doc/colortail/examples/ .
In figure 1 you can see an example output of colortail on the syslog of a virtual machine.
multitail
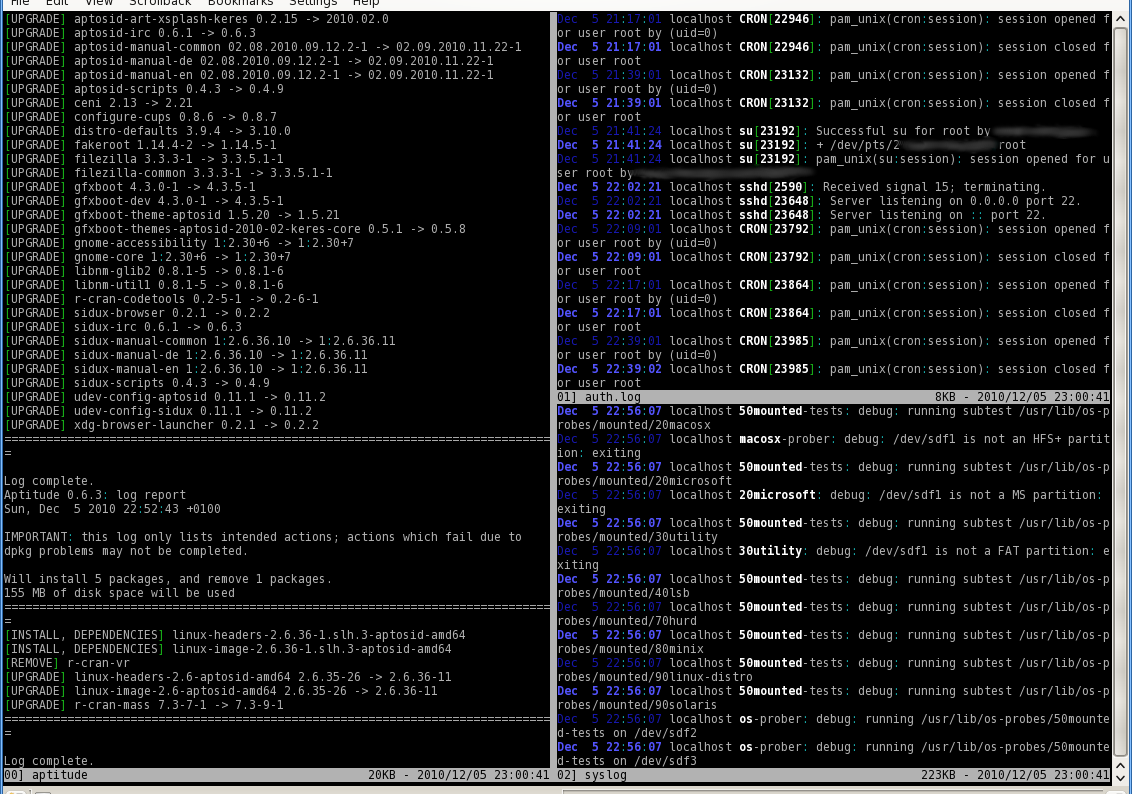
The second tool in this article is multitail. Like colortail it can colorize the output, but all is presented in a ncurses based user interface so it is able to create multiple windows on your console. If you open a file in multitail it’s automatically in a following mode ( -f in case of tail and colortail).
If you are monitoring multiple log files your console is split horizontal or vertical or a mix of both. You can pause the output, search for regular expressions and a lot more. Enter F1 to get a small help window.
Figure 2 presents a sample output. Its project page keeps much more information.
logtail
logtail pursues a different goal. It’s not interested in prettifying the output, it remembers the content that was still displayed and just prints the differences to the last run. So it is an ideal tool for log analyzer, log messages doesn’t have to be parsed multiple times. logtail is written in perl, you can also monitor logfiles on different machines.
I hope I could give you some smart inspirations.
OpenNIC DNS network
December 3rd, 2010DNS look-ups are a very sensible topic. Of course you want very fast name-to-IP resolutions, but should you always use Google’s DNS server? After all they can keep track of all your network motion profile unless you are surfing by IP! Today I read about the OpenNIC Project and ran some speed tests. It’s very interesting and worthy to know about!
The project about itself:
OpenNIC (a.k.a. "The OpenNIC Project") is an organization of hobbyists who run an alternative DNS network. [...] Our goal is to provide you with quick and reliable DNS services and access to domains not administered by ICANN.
Ok, I gave it a try and implemented a Perl-script that checks the speed. It throws a dice to call one of my often used domains and digs1 each of my predefined DNS servers to save the query time. I tested the following DNS server:
178.63.26.173: one server of the OpenNIC project, located in Germany217.79.186.148: one server of the OpenNIC project, located in Germany (NRW)8.8.8.8: Google’s public DNS server, proven to be fast and reliable172.16.20.53: my ISP’s server141.48.3.3: name server of our university
Find the Perl code attached.
And here are the results after 10000 qeuries:
| IP | Provider | 10000 queries |
|---|---|---|
| 172.16.20.53 | my ISP | 131989 ms |
| 217.79.186.148 | OpenNIC | 259382 ms |
| 8.8.8.8 | 270300 ms | |
| 178.63.26.173 | OpenNIC | 304094 ms |
| 141.48.3.3 | NS of uni-halle.de | 394134 ms |
As you can see, my ISP’s DNS server is the fastest, they may have optimized their internal infrastructure to provide very fast look-ups to its customers. But it is also nice to see, that there is one OpenNIC server that is faster than google! And this server comes with another feature: It doesn’t track any logs! Isn’t that great!?
To find some servers near you just check their server list. Some of them don’t record logs or anonymize them, and of course all of them are independent from ICANN administrations.
I can’t recommend to use any special DNS server, but I want to advise to test them and find the best one for your demands! Feel free to post your own test results via comment or trackback.
1 dig is part of the larger ISC BIND distribution
Boot messages to service console
December 1st, 2010You may have heard about management consoles!? If a server is dead you can revive it via service console without driving the long way to the data center (often miles away).
While logged into the service console you of course have the chance to reboot the machine itself. To get to know what it is doing while booting you may want to see all the messages that are usually prompted to the terminal at the attached monitor. Unfortunately you aren’t next to the machine, and so there is no monitor attached to it, but you can force grub to prompt all messages both to terminal and to service console.
First of all you have to setup the serial console:
serial --unit=0 --speed=57600 --word=8 --parity=no --stop=1The --unit parameter determines the COM port, here it’s COM1, if you need COM2 you should use --unit=1 . --speed defines a baud rate of 57600 bps, see your manual. To learn more about the other parameter you are referred to the Grub manual for serial.
Next you have to tell Grub where to write the output:
terminal --timeout=5 console serialThis line tells grub that there are two devices, the typical console on the attached screen and our previous defined serial console. With this directive Grub waits 5 seconds for any input from serial console or the attached keyboard and will print its menu to that device where the input was generated. That means if you’re at home and press any key, Grub will show you all outputs to your serial connection, but your student assistant (who had to go to the server, by bike while raining!!) isn’t able to see whats happening. But if your assistance is faster than you and hits a key on the physically attached keyboard, he’ll see anything and you’ll look through a black window… If nobody produces any input the output is written to that device that is listed first.
Last but not least you have to modify the kernel sections of the boot menu and append something like that at the end of every kernel line:
console=tty0 console=ttyS0That tells grub that all kernel messages should be printed to both the real console of the attached screen and the serial console. Keep in mind to modify ttyS0 to match your serial port (here it is COM1).
Grub decides for the device that is listed last to also send all stdin/stdout/stderr of the init process, that means only the last device will act as interactive terminal. E.g. checks of fsck are only printed to the last device, so stay calm if nothing happen for a long time on the other one ;-)
Here is a valid example for copy and paste:
# init serial console
serial --unit=0 --speed=57600 --word=8 --parity=no --stop=1
# what device to use for grub menu!?
terminal --timeout=5 console serial
# ....
title Debian GNU/Linux, LOCAL CONSOLE
root (hd0,0)
kernel /vmlinuz-SOMEWHAT-openvz-amd64 root=UUID=AAAAAAAA-BBBB-CCCC-DDDD-EEEEEEEEEEEE ro console=ttyS0 console=tty0
initrd /initrd.img-SOMEWHAT-openvz-amd64
title Debian GNU/Linux, LOCAL CONSOLE
root (hd0,0)
kernel /vmlinuz-SOMEWHAT-openvz-amd64 root=UUID=AAAAAAAA-BBBB-CCCC-DDDD-EEEEEEEEEEEE ro console=tty0 console=ttyS0
initrd /initrd.img-SOMEWHAT-openvz-amd64Here both Grub entries are booting the same kernel, but the first one will use the local console as interactive terminal whether the other entry takes the serial console for interactions.
